Exploring Nanosecond Motions at Atomic Resolution in Proteins with High-Resolution Relaxometry
Prof. Fabien Ferrage
Internal motions in proteins are essential for their functions, but how do proteins move? NMR relaxation is an invaluable method to probe local motions in proteins [1]. NMR relaxation in proteins has been measured and analyzed for several decades. The most popular approach consists in recording nitrogen-15 relaxation rates in an isotopically labeled protein and analyze these rates with the model-free formalism [2]. One obtains order parameters that describe the equilibrium distribution of orientations and an effective timescale for local motions.
Internal motions in proteins are essential for their functions, but how do proteins move? NMR relaxation is an invaluable method to probe local motions in proteins [1]. NMR relaxation in proteins has been measured and analyzed for several decades. The most popular approach consists in recording nitrogen-15 relaxation rates in an isotopically labeled protein and analyze these rates with the model-free formalism [2]. One obtains order parameters that describe the equilibrium distribution of orientations and an effective timescale for local motions.
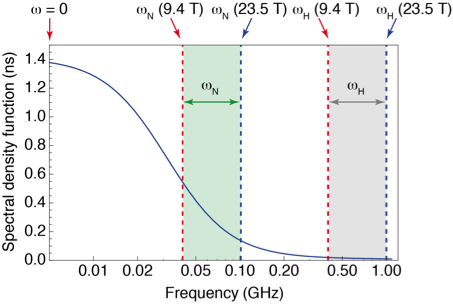
However, high-field measurements of relaxation only probe motions at the resonance frequencies of the system, leaving broad gaps where no information is gathered (see Fig. 1). To fill these gaps, relaxation measurements have to be performed at lower magnetic fields, where the sensitivity and resolution are insufficient for protein NMR. Several groups (R. G. Bryant, A. G. Redfield, H. M. Vieth, K. L. Ivanov, T. H. Huang) and companies (Field Cycling Technology Ltd., Bruker Biospin) have worked on a technological solution: designing fast sample shuttle systems that make it possible to measure relaxation at low fields in the stray field of a commercial magnet, while polarization and detection take place at high field.
The first investigation of site-specific dynamics in a protein by high-resolution relaxometry was performed by the groups of Al Redfield and Dorothee Kern [5]. Using the sample shuttle system built by Al Redfield[6], they measured 15N longitudinal relaxation rates down to 4 T (17.3 MHz resonance frequency for 15N) on the nucleocapsid protein of virus SARS-CoV (before there was a need to give a number to the SARS-CoV…). The motion of an entire β-haipin was identified, with an effective correlation time around 0.8 ns.
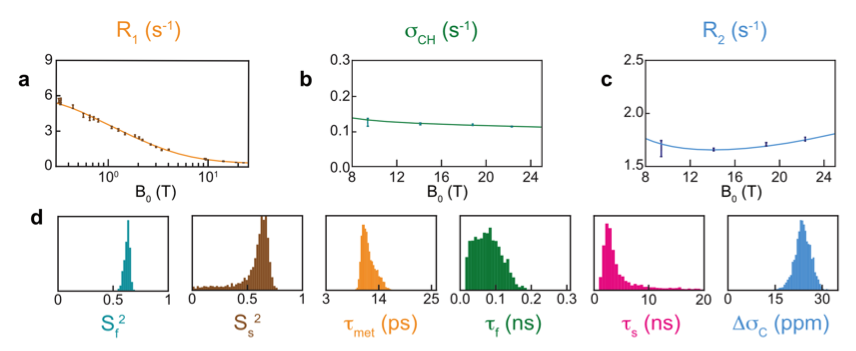
We have investigated site-specific motions on nanosecond timescales in the protein ubiquitin using a prototype developed by Bruker Biospin [7]. In particular, we have recently investigated motions in methyl-bearing side chains through measurements of 13C relaxation in 13C1H2H2 methyl groups from 0.33 T to 22.3 T. We found, for 3 out of 7 isoleucine sidechains, that significant motions on low nanosecond timescales could be identified from relaxometry data[3].
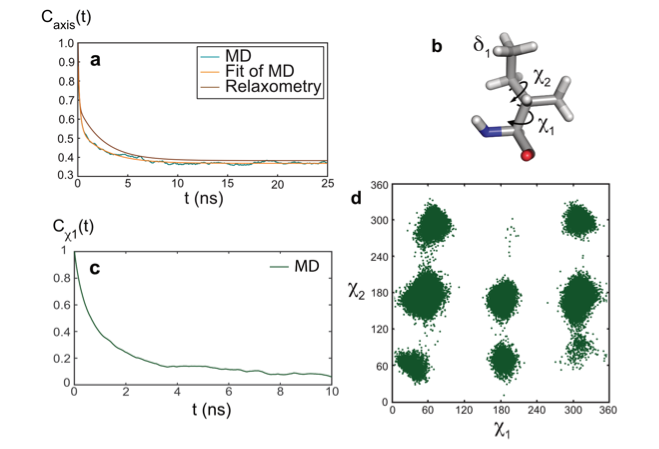
The model-free analysis of such relaxation datasets is convenient and instructive, yet, it does not provide a mechanistic description of the motions detected. To gain additional insight into the nature of these motions, the combination of molecular dynamics (MD) simulations and experimental approaches is essential. The results of the analysis of relaxometry and MD simulations (carried by the group of Rafael Brüschweiler) agreed well for the side chain of isoleucine 36 in ubiquitin, which gave us an opportunity to explore the nature of nanosecond motions. In this case, nanosecond motions are dominated by the dynamics of rotamer jumps around the χ1 dihedral angle.
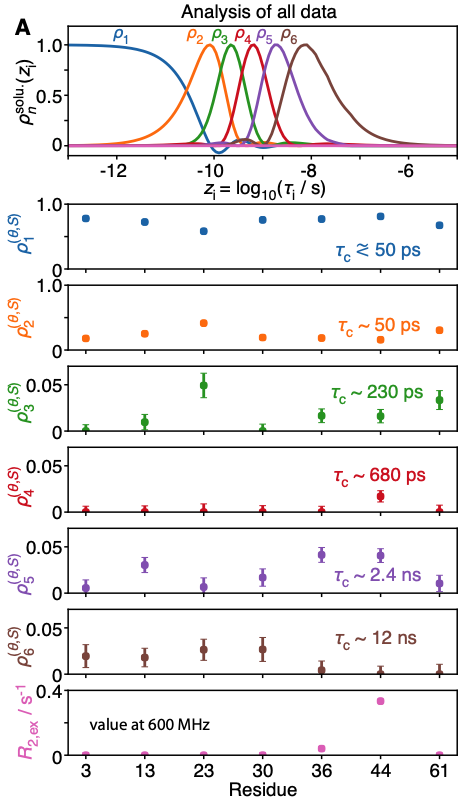
A recent investigation of this set of thirty relaxation rates per methyl group, based on the detectors approach [8], confirmed that low-field relaxation rates were essential to characterize the nanosecond motions of two of these methyl groups: those of Ile13 and Ile36, that are characterized by effective correlation times between 2 and 4 ns [4]. Importantly, the optimization of detectors for small and medium-sized proteins highlights the increase of resolution expected in the determination of slow, nanosecond-timescale motion achieved by high-resolution relaxometry, in particular for larger proteins, showing the way for further investigations.